

-
Ultrafast excited-state dynamics of a series of zwitterionic pyrydinium phenoxides with increasing sterical hindering
G. Duvanel, J. Grilj, H. Chaumeil, P. Jacques and E. Vauthey
Photochemical and Photobiological Sciences, 9 (2010), p908-815


DOI:10.1039/c0pp00042f | unige:14768 | Abstract | Article HTML | Article PDF
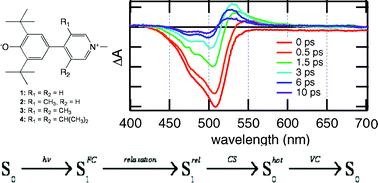
A series of pyridinium phenoxides that differ by the dihedral angle between the pyridinium and the phenoxide rings because of substituents with increasing steric encumbrance has been investigated by ultrafast spectroscopy. Like the related betaine-30, these molecules are characterised by a zwitterionic electronic ground state and a weakly polar S1Â state. Their fluorescence lifetime was found to lie between 200 to 750 fs, decreasing with increasing dihedral angle, and increasing with solvent viscosity. This was assigned to a non-radiative deactivation of the emissive state coupled to a large amplitude motion involving the dihedral angle. The transient absorption spectra suggested that emission occurs from the FranckâCondon S1Â state, which decays to a dark excited state, that itself most probably corresponds to the relaxed S1Â state. Finally, this relaxed state decays to the vibrationally hot ground state through an intramolecular charge separation process with a time constant ranging between 0.4 and 3 ps, increasing with the dihedral angle and with the solvent relaxation time. These variations were discussed in terms of the JortnerâBixon model of electron transfer, where the charge separation dynamics depends on both electronic coupling and solvent relaxation. The results suggested that charge separation slows down with increasing dihedral angle.